1 Introduction
Chronic Obstructive Pulmonary Disease (COPD) is a term used to describe various lung diseases characterized by airflow limitation, including emphysema, chronic bronchitis, refractory (non-reversible) asthma, and certain forms of bronchiectasis. COPD is one of the leading causes of death worldwide, causing an estimate of 3 million deaths in 2015, and is hypothesized to be associated with heritable genetic factors as well as environmental exposures (e.g., smoking).
One of the conventional ways to uncover genes associated with diseases is through a genome-wide association study (GWAS). This method examines the whole genome to find positions (loci) where patients suffering from COPD (cases) and those who do not (controls) differ significantly in nucleotides proportion, suggesting that it may influence the risk of the disease.
Through GWAS, 836 loci have been discovered to be associated with COPD [1],[2]. However, these discovered loci do not provide information on how these loci affect lung function; for example, these loci may regulate the expression of the closest gene, or it could be distantly affecting other genes. For more insight, we can examine transcriptome data, which provides information on levels of messenger RNA that were transcribed from genes. The challenge with this approach is that there is a smaller number of individuals with observed transcriptome data than genotype data. To overcome these challenges, this project seeks to identify COPD-associated genes by looking at the predicted gene expression that was calculated from the larger genotype dataset.
In this project, we explored different association tests to find genes associated with COPD. We used a novel statistical test, developed by Aziz Mezlini, one of the co-authors of this paper.
2 Approach
We were provided with transcriptome and genotype datasets from populations of two ethnicities: African-American and Non-Hispanic White.

Figure 1:Number of individuals in given dataset. A Comparison between the total number of individuals with genotype and transcriptome data. B The exact numbers of individuals in dataset grouped by ethnicity and status
The core of the approach was to apply machine learning methodologies to predict the genetically-regulated component of the gene expression; this was achieved by incorporating regions of the genome which have been known to influence the expression level of one or more genes, regions known as expression quantitative trait loci (eQTL). These eQTL regions serve as our predictor variables in the predictive model. There have been two new tools that attempt to predict gene expression using eQTL, namely PrediXcan [3] and fQTL [4]. These two tools approach the problem from a slightly different angle; while PrediXcan adopts an elastic net model, training on different tissue type datasets separately, fQTL takes a multi-tissue, multivariate approach, in which the eQTL effects are decomposed into tissue-specific and loci-specific components. For this project, we predicted gene expression using PrediXcan and fQTL for whole-blood and lung tissues.
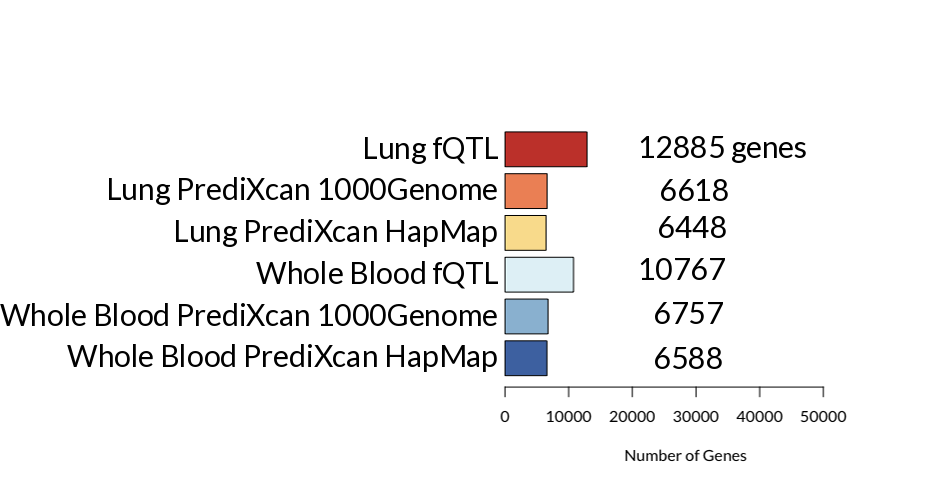
Figure 2: The number of genes predicted by fQTL and PrediXcan models.
After predicting gene expression using PrediXcan and fQTL, we performed association tests through different hypothesis-driven approaches, integrating transcriptome information with predicted expression.
We used the following statistical tests during association analysis:
- T-test
- Logistic regression of gene expression values on case/control status
- Logistic regression of gene expression values on case/control status and smoking duration
- Mezlini’s test
Mezlini’s test is a new statistical test developed by Aziz Mezlini to analyze if two populations are different by looking for enrichment close to the end of a distribution, as depicted by Figure 3.
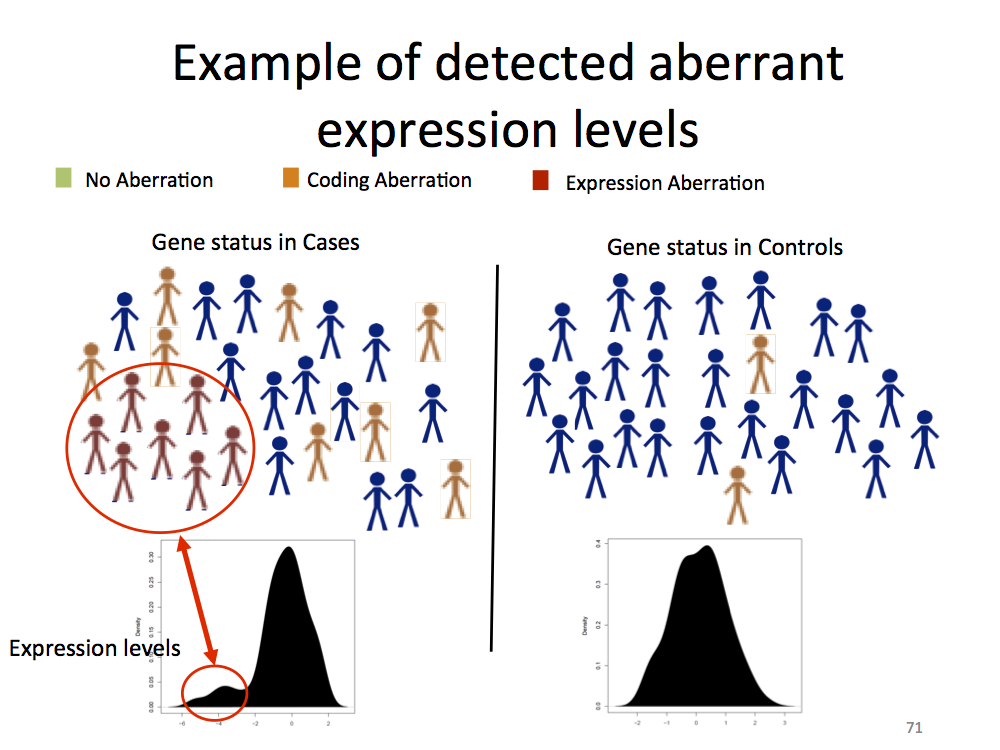
Figure 3: Mezlini’s test looks for enrichment at the tail of a distribution. This figure depicts distribution of cases (target phenotype) and controls, where it is deemed as not associated by t-test but deemed as associated by Mezlini’s test.
We identified 15 statistically-significant genes, 2 of which were previously discovered by GWAS. These genes genetically interacted and are located in the vicinity of previously GWAS-discovered loci.
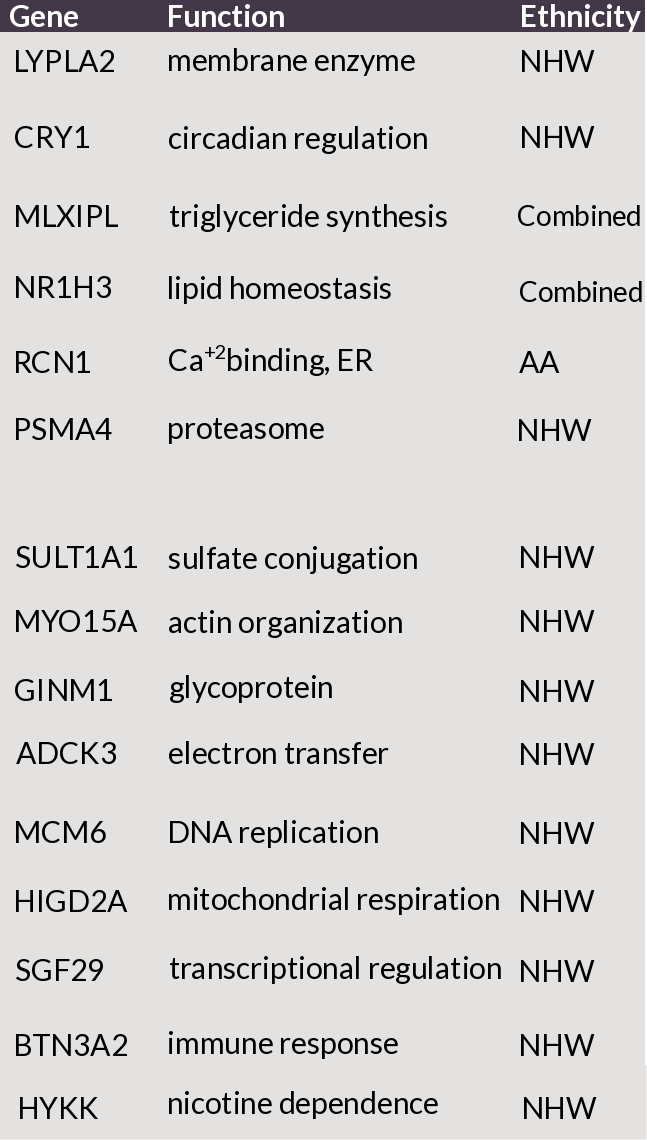
Figure 4: List of genes discovered to be associated with COPD with its function and ethnicity. HYKK and PSMA4 were previously reported (by GWAS) to be associated with COPD.
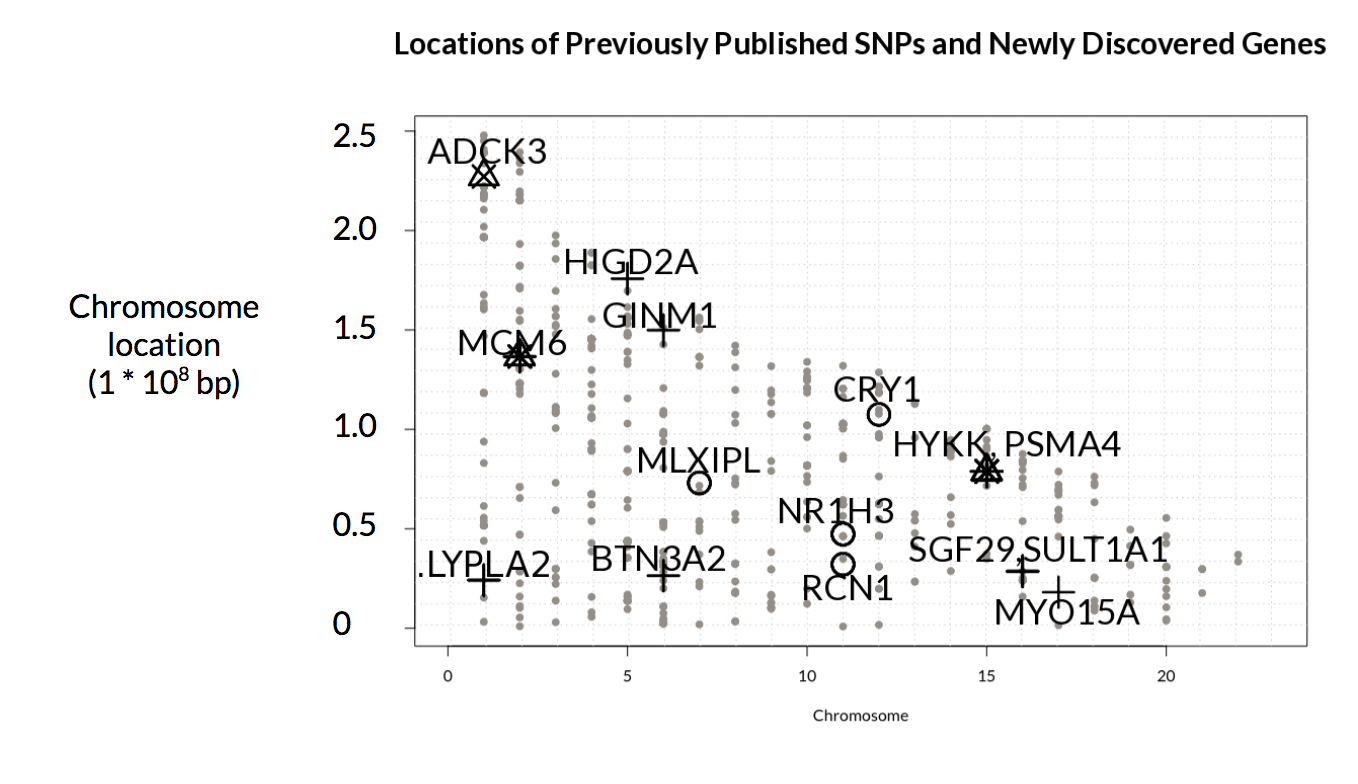
Figure 5: Locations of previously published loci (in grey) and discovered genes (in black). As can be seen from the figure, the discovered genes are located near previously published loci. We can also observe that Mezlini’s test discover genes overlooked by T-test and logistic regression. Conclusion.
Through this project, we have shown that Mezlini’s test is able to detect associations overlooked by other statistical tests, as shown in Fig. 5. Notably,, we have also found different genes associated with COPD in African-American and Non-Hispanic White datasets. This may be due partly to the difference in the numbers of individuals (AA:NHW = 2801 :5982). Additionally, it suggests that genes have varying degrees of predisposition (to COPD) among ethnicities.
This project also shows that GWAS is insufficient in providing insight on the genetics behind COPD; inspecting predicted expression provides such insight on how these genes influence certain traits.
Acknowledgements
Thank you to Dr. Anna Goldenberg and Aziz Mezlini for the continuous guidance and support! Special thanks to our collaborators from Brigham and Women’s Hospital, Harvard Medical School who have provided us with the COPD datasets: Michael Cho and Craig Hersh.
Shoutout to all the lab members for all the help, discussions, and brainstorming sessions: Ben, Lauren, Daniel H., Ladislav, Shems, Mingjie, Kingsley, Daniel C., Lebo, Jaryd, Mohamed, and Walter.
Citations
===
- K. Probert et al., “Developmental genetics of the COPD lung,” COPD Research and Practice, vol. 1, no. 1, pp. 10, Nov. 2015. doi: 10.1186/s40749-015-0014-x
- T. Burdett et al. (2017, August 14). The NHGRI-EBI Catalog of published genome-wide association studies [Online]. Available: http://www.ebi.ac.uk/gwas
- E. Gamazon et al., “A gene-based association method for mapping traits using reference transcriptome data,” Nat. Gen., vol. 47, no. 9, pp. 1091-1098, Sep. 2015. doi: 10.1038/ng.3367
- Y. Park et al., “Multi-tissue polygenic models for transcriptome-wide association studies,” bioRxiv, 2017. doi: 10.1101/107623